El síndrome de Rett és una malaltia del desenvolupament neurològic que constitueix la segona causa més freqüent de retard mental en dones, després de la Síndrome de Down.
El quadre clínic comença a aparèixer 6-18 mesos després del naixement i consisteix en una pèrdua de capacitats cognitives, socials i motores acompanyada de comportaments autístics com, per exemple, moviments esterotipats de les mans. Avui dia no existeix tractament efectiu de la malaltia més enllà del control
de la seva simptomatologia.
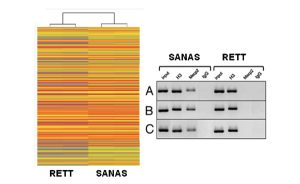
La síndrome sol ser a causa de la presència d’una mutació al gen MeCP2, un gen epigenètic que com si fos un imant regula l’expressió de molts altres gens de la cèl·lula. No obstant això, encara hi ha un gran desconeixement de les alteracions moleculars presents a la malaltia. El grup del Dr Manel Esteller, Director del Programa d’Epigenètica i Biologia del Càncer de l’Institut de Investigacions Biomedicas de Bellvitge, Investigador ICREA i Professor de Genètica de la Universitat de Barcelona, descriu aquesta setmana a la revista biomèdica internacional RNA Biology la identificació de fils especials de àcid ribonucleic (ARN) alterades a la Síndrome de Rett denominades ARNs no codificants de llarga cadena (lncARNs). Els lncARNs actuarien com agents supervisors encarregats d’apagar o encendre altres gens de el nostre genoma i així regular l’activitat de les neurones.
Els investigadors van recórrer a un model de raó que reproduïa realment les característiques de la síndrome de Rett humà i en el mateix compararon l’expressió de les cadenes llargues de ARN en animals sans y malalts. El grup liderat pel Dr Manel Esteller ha descobert que la presencia de mutacions en el gen Mecp2 provocava alteracions en la activitat dels ARN llargs no codificats originats a partir del “genoma oscuro”, aquell on no es troben els gens típics ni produeixen proteïnes.
Entre els lncARNS identificats com a alterats a les regions cerebrals afectades en la síndrome de Rett es troba un que regula la funció del receptor GABA. Esta ultima molecula (cuyas sigles es refereixen a l’àcid gamma- aminobutírico) és un neurotransmisor clau en el sistema nervioso cerebral de tots els vertebrats i la seva alteració podrien explicar els defectes de comunicació neurona-neurona a les nenes afectades per la síndrome de Rett. Aquests descobriments, a més d’augmentar el coneixement sobre les causes d’aquesta malaltia, podria tenir conseqüències futures per al tractament d’aquests pacients ja que teràpies que tuvieran com diana els lncARNs o el receptor GABA mereix ser estudiades en assaigs preclínics.
L’estudi ha estat recolzat pel Departament de Salut de la Generalitat de Catalunya, la Institució Catalana d’Estudis Avançats (ICREA), el Ministerio
de Sanidad y Consumo (E-RARE), los Proyecto Europeos DISCHROM y EPINORC, la Fondation Lejeune (Francia) i l’Associació Catalana del Síndrome de Rett.
Article: Desregulació del transcriptoma llarg d’ARN no codificant en un Rett model de ratolí de síndrome. Petazzi P, Sandoval J, Szczesna K, Jorge OC, Roa L,
Sayols S, Gomez A, Huertas D, Esteller M. RNA Biology, 10(7), 2013.
Deja una respuesta